Sickle Cell Disease
Congenital haemoglobinopathy leading to production of the relatively insoluble haemoglobin S, causing erythrocytes to adopt a ‘sickle’ shape under conditions of physiologic stress. Sickle cell anaemia leads to:
- Vaso-occlusion and haemolysis
- Pain due to microvascular obstruction
- Progressive multiorgan dysfunction due to ischaemia
- Relative protection against malaria
Sickled cells haemolyse prematurely, interrupting the Plasmodium life-cycle.
Epidemiology and Risk Factors
Pathophysiology
Autosomal recessive:
- Homozyogotes have sickle cell disease
- Chronic haemolytic anaemia
- Recurrent vaso-occlusion and severe pain
- Progressive organ damage
- Often early death
- Heterozygotes have sickle cell trait
- Results in HbS levels of ~30-40%
- HbS may precipitate in conditions of physiologic stress
- Usually well
HbS:
- Work normally when oxygenated
- Deforms in hypoxia, resulting in polymerisation and deformation of the red cell into a sickle shape
Sickled cells:- Are rigid and sticky
Can obstruct capillary flow, leading to vaso-occlusion. - Haemolyse rapidly
Lifespan of 10-20 days- Leads to anaemia due to rapid turnover
- Are rigid and sticky
Aetiology
Clinical Manifestations
Sickle cell anaemia may present as a series of different crises.
Vasoocclusive crisis:
- Sudden onset pain
Ischaemia or infarction secondary to microvascular obstruction. - Hypovolaemia
Inability to concentrate urine.- Generally hypernatraemic, and require volume resuscitation with hypotonic solutions
Sequestration crisis:
- Pooling of erythrocytes in spleen and liver
Presents as:- Anaemia
- Hypovolaemic shock
Assessment
History:
Exam:
Investigations
Bedside:
Laboratory:
- Blood
- FBE
- Chronic anaemia
- Hb electrophoresis
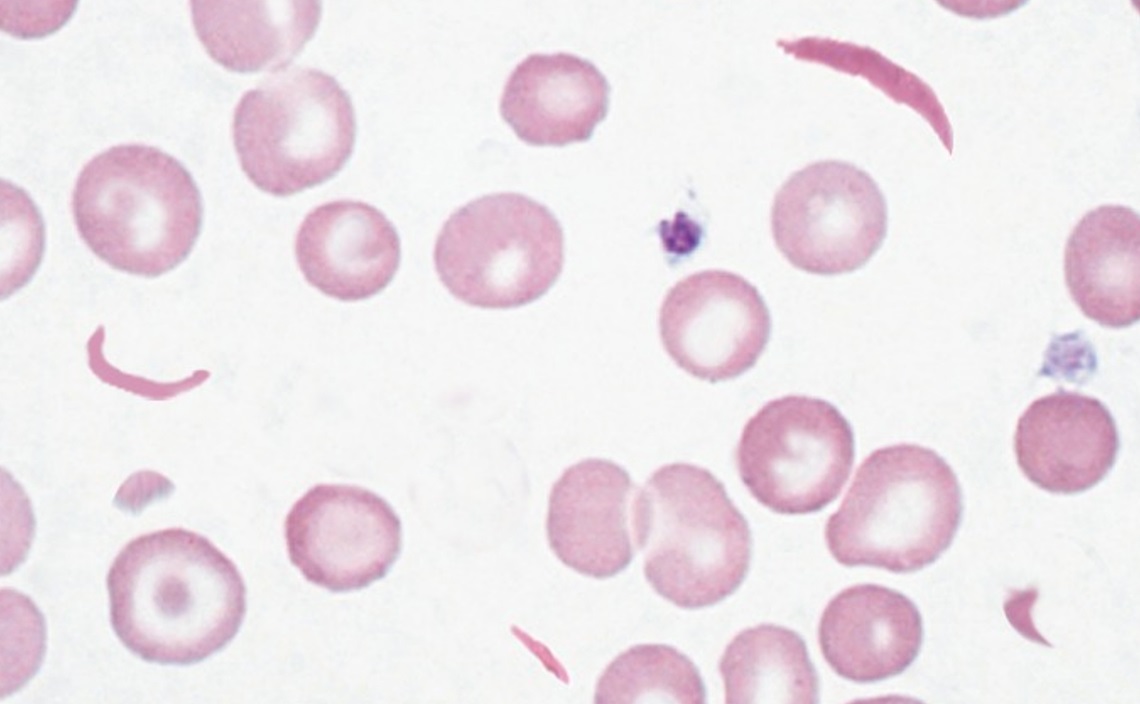
For diagnosis. - Blood film:
- Sickle forms
- Reticulocytosis
- Sickledex test:
- Rapid screening
- Qualitative detection of HbS levels >10%
- FBE
Imaging:
Other:
Diagnostic Approach and DDx
Management
- Prevent and treat sickling precipitants
- Transfusion
Resuscitation:
Specific therapy:
- Pharmacological
- Vaccination
If asplenic.
- Vaccination
- Procedural
- Bone marrow transplantation
Usually restricted to <16 year olds. - Exchange transfusion
May be required for severe vasoocclusive crises.
- Bone marrow transplantation
- Physical
Asplenism is covered under Asplenia.
Supportive care:
- H
- Hb >50
Disposition:
Other:
Prevention:
- Avoid precipitants
- Dehydration
- Exercise
- Hot weather
- Alcohol
- Hypoxia
- Smoking
- OSA
- Physiological stress
- Hypothermia
- Dehydration
- Folic acid supplementation
- Hydroxyurea
↑ HbF formation, and so reduces frequency and severity of vaso-occlusive crises.
Medical
Surgical
Anaesthetic Considerations
Avoid precipitants of vaso-occlusive crises:
- Hypoxia
- Acidosis
- Hypercapnoea
- B
- PHTN
- C
- CCF
- Volume state
- Minimise fasting period
- Preoperative hydration
- Avoid venous stasis
- E
- Tourniquet use
- May precipitate sickle crisis distal to tourniquet
- Careful exsanguination of distal limb prior to inflation
- May precipitate sickle crisis distal to tourniquet
- Tourniquet use
- H
- Sickle cell severity
- Frequency of exacerbations
- Hb
- Should be transfused to Hb >100g/dL or HbS <30%
- Sickle cell severity
Marginal and Ineffective Therapies
Complications
- B
- Pulmonary hypertension
Recurrent pulmonary infarction.
- Pulmonary hypertension
- C
- Cardiac failure
- D
- TIAs/CVAs
- H
- Functional asplenism
Consequential lymphoid hypertrophy.
- Functional asplenism
Prognosis
Key Studies
References
- Wilson M, Forsyth P, Whiteside J. Haemoglobinopathy and sickle cell disease. Contin Educ Anaesth Crit Care Pain. 2010;10(1):24-28. doi:10.1093/bjaceaccp/mkp038