Haemophagocytic Lymphohistiocytosis
Life-threatening hyperimmune state characterised by excessive macrophage and T-cell activation secondary to ↑ cytokine production, which may be either:
- Congenital
- Acquired
Relatively underdiagnosed, precipitants include:- Infection
- Malignancy
- Rheumatological disease
Also known as macrophage activation syndrome in this context.
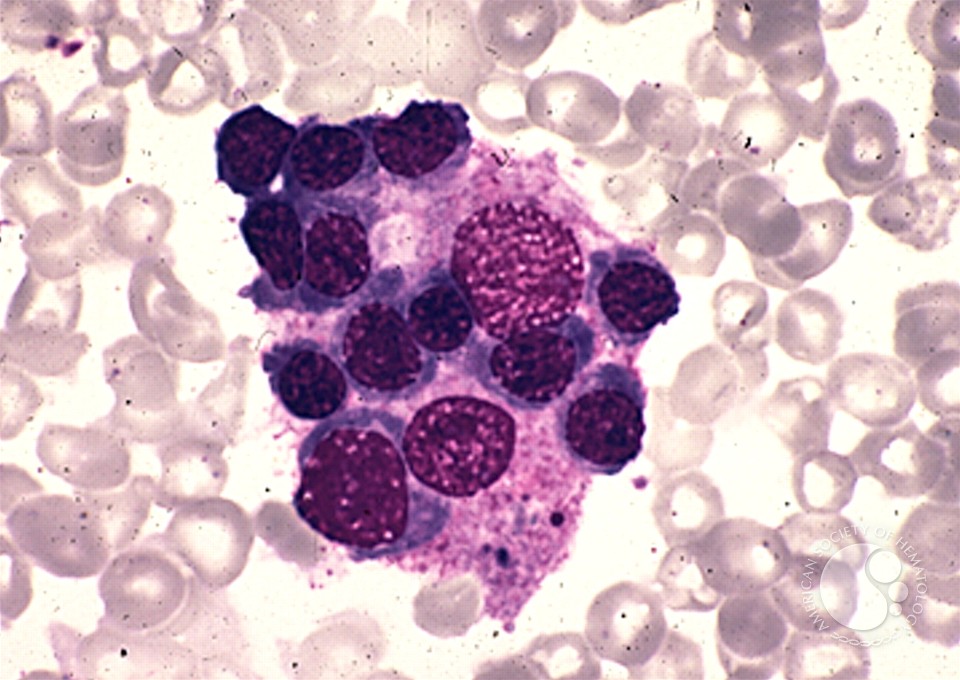
The name comes from the blood film findings which demonstrate macrophages ingesting other blood cells.
The division between congenital and acquired forms are becoming blurred; as there is ↑ recognition of the interplay of genetic, disease, and environmental factors that may lead to different clinical disease expression. That aside, for practical purposes the information here refers to adult expressions HLH.
Epidemiology and Risk Factors
Pathophysiology
Two complementary mechanisms:
- Loss of inflammatory negative feedback mechanisms
Genetic mutation in genes that prevent positive feedback loops occurring in inflammation.- Primary determinant in paediatric presentations
- Less important in adult HLH
- Inflammatory trigger
- Infection
- Malignancy
Aetiology
Causes include:
- Infection
- Malignancy
- Haematological malignancy
- Lymphomas
- Haematological malignancy
- Transplantation
- Solid organ
- Bone marrow transplant
- Immunotherapy
- CAR T-cell therapy
| Viral | Bacterial | Fungal | Parasitic |
|---|---|---|---|
|
|
|
|
EBV may be a direct trigger, or cause HLH via an associated malignancy.
Clinical Features
- CNS
25-50% of cases, may rarely occur without systemic features:- Delirium → obtundation → coma
- Focal deficits
- Headaches
- Meningism
- PRES
- Haematological
- Splenomegaly
- Hepatomegaly
- Petechiae → generalised purpura
- Inflammatory
- Fever
Classically persistent, non-remitting, and non-responsive to antibiotics. - Capillary leak
- Rashes
- Lymphadenopathy
- Myocarditis
- ARDS
- AKI
- Fever
Assessment
History:
Exam:
Investigations
Bedside:
Laboratory:
- Blood
- Diagnosis
- FBE
Cytopenias. - Iron studies
- Soluble CD25
Indicates T-cell activation. - Blood film
- FBE
- Complications
- LFTs
Hepatitic picture. - ↑ Triglycerides
- Coagulation abnormality
Hyperfibrinolysis from macrophage activation.
- LFTs
- Precipitant
- HIV
- ANA
- Viral PCR
- (1→3)-β-D-glucan
- Diagnosis
Imaging:
- CT
For malignancy or occult infection.
Other:
- Bone marrow biopsy
- Repeat biopsy may be required
Insensitive in early disease.
- Repeat biopsy may be required
- Lumbar puncture
- Primary indication is exclusion of alternative diagnoses
- Must be balanced against any risk with coagulopathy
- May demonstrate haemophagocytosis
Diagnostic Approach and DDx
Congenital HLH is diagnosed on genetic testing. Acquired HLH requires five of:
Genes which may accrue mutations consistent with HLH include: PRF1,UNC13D, Munc18-2, Rab27a, STX11, SH2D1A, or BIRC4.
- Fever
- Splenomegaly
- Poly-cytopenia
⩾2 cell lines. - Histological evidence of haemophagocytosis
- Ferritin >500μg/mL
May be substantially higher:- >3000μg/mL is concerning
- >10,000μg/mL is 96% specific and 90% sensitive in children
- Low or absent NK activity
- Soluble IL-2 receptor CD25 >2400U/mL
The utility of this is questionable in practice given the long turna round time.
There is a substantial overlap in features with sepsis (see Sepsis); consider HLH as a differential diagnosis in septic patients without a source.
Implementing these criteria disease is hard in practice:
- Haemophagocytosis is insensitive early in disease
- NK-cell activity and CD25 have a long latency before they return
- Critically ill patients are critically ill, and require immediate management
Management
- Treat underlying disease
- Immunosuppression
- Corticosteroids
- Anakinra
Resuscitation:
Specific therapy:
- Pharmacological
- Corticosteroids
- Daily steroid
- Dexamethasone 15-20mg IV daily
Preferred in neurological disease due to CNS penetration.
- Dexamethasone 15-20mg IV daily
- Pulsed steroids
Typically preferred for rheumatological disease:- Methylprednisolone 1g IV daily for 3 days, then 2-3mg/kg/day
- Daily steroid
- Anakinra 1-2mg/kg SC Q12H
Preferable to steroids inf the diagnosis is unclear. - IVIG 2g/kg
- Later-line therapies
Discuss with a haematologist:- Cyclosporin
- Etoposide
- Corticosteroids
- Procedural
- Bone marrow transplant
Considered in selected cases.
- Bone marrow transplant
- Physical
Anakinra is a recombinant IL-1 receptor antagonist that ↓ the cytokine storm.
Supportive care:
Disposition:
Preventative:
Marginal and Ineffective Therapies
- Plasma exchange
Anaesthetic Considerations
Complications
- Death
40%; ↑ risk with:- Neurological symptoms
- Ferritin >5,000μg/mL
- H
- DIC
Prognosis
Key Studies
References
Procedure
Indications
Contraindications
Anatomy
Equipment
Technique
Complications
References
- Bauchmuller K, Manson JJ, Tattersall R, et al. Haemophagocytic lymphohistiocytosis in adult critical care. Journal of the Intensive Care Society. 2020;21(3):256-268. doi:10.1177/1751143719893865